
Researchers
Maurizio Molinari – Group Leader
Tatiana Soldà – Scientist
Status: in progress
Overview
The aim of the project is to identify new players that intervene in protein folding, quality control and ERAD in the mammalian ER lumen. We generated a collection of human cell lines expressing epitope-tagged folding-competent and folding-defective proteins. The model proteins are used as baits to capture interacting partners in the same immuno-complexes. The proteins co-immunoisolated with the individual baits are subjected to tryptic digestion and fragments are separated by nano-HPLC followed by tandem mass spectrometry. Fragmentation spectra of the samples are matched to a human protein database sequence with the Mascot software. These analytic steps are performed in collaboration with Manfredo Quadroni, coordinator of the Center for Integrative Genomics, University of Lausanne. Involvement in protein quality control of the interacting partners of the model proteins will be validated in 2 steps: i) confirmation of interaction by co-immunoisolation followed by western blot; ii) evaluation of the role of the interactors by monitoring consequences on the substrate fate upon silencing of their expression or upon co-expression with the model substrate of their dominant negative mutants.
Researchers
Maurizio Molinari – Group Leader
Tatiana Soldà – Scientist
Elisa Fasana – Scientist
Status: in progress
Overview
To maintain ER homeostasis and to ensure the highest efficiency of functional polypeptide production, the quality control machinery operating in the mammalian ER must distinguish non-native intermediates of protein folding programs from terminally misfolded polypeptides. Folding intermediates must be retained in the ER to attain the native structure under the assistance of dedicated molecular chaperones and folding enzymes. Terminally misfolded polypeptides must be rapidly cleared from the ER lumen to avoid interferences with ongoing folding programs. For glycosylated polypeptides, which represent the majority of the cargo entering the secretory pathway, the processing of the N-linked oligosaccharides determines retention in the folding environment (cycles of removal/re-addition of terminal glucose residues) or extraction from the folding environment for disposal. Virtually nothing is known about quality control of non-glycosylated polypeptides. The aim of this project is the identification of ER-resident factors involved in quality control and disposal of both soluble and membrane-bound non-glycosylated variants of model glycopolypeptides generated in our lab.
Publications
N-linked sugar regulated protein folding an quality control in the ER
Tannous et al.,
in Semin Cell Dev Biol. 2015, 41:79-89
Researchers
Maurizio Molinari – Group Leader
Ilaria Fregno – Research Assistant
Elisa Fasana – Research Assistant
Marika Kucińska – PhD Student
Mikhail Rudinskiy – PhD student
Status: in progress
Overview
ER-phagy defines the constitutive and regulated clearance of ER portions by endolysosomes or vacuoles. It is induced by various cues such as nutrient deprivation, perturbation of calcium, sugar or redox homeostasis, ribosome stalling, pathogen invasion, accumulation of misfolded polypeptides, formation of ER whorls. It proceeds via at least three distinct pathways (i.e., macro-ER-phagy, micro-ER-phagy and LC3/Atg8-dependent vesicular transport) that rely on engagement of specific autophagy, trafficking, membrane remodelling, membrane fusion, chaperone, hydrolytic gene products. Specificity of the pathways is determined by ER-phagy receptors, i.e., LC3/Atg8-binding protein that span or peripherically associate with the ER membrane. A major activity of our group aims at understanding the molecular mechanisms that regulate ER-phagy pathways in mammalian cells.
Publications
ER-phagy: mechanisms, regulation and diseases connected to the lysosomal clearance of the endoplasmic reticulum
Reggiori, F. Molinari, M.
in Physiol Rev (2022) Vol. pp
N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation
Fregno, I. Fasana, E. Solda, T. Galli, C. Molinari, M.
in EMBO J (2021) Vol. ppe107240
Endoplasmic Reticulum (ER) and ER-Phagy
Loi, M. Marazza, A. Molinari, M.
in Prog Mol Subcell Biol (2021) Vol.59 pp99-114
ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD
Molinari, M.
in Dev Cell (2021) Vol.56 pp949-966
ER-phagy: Eating the Factory
Molinari, M.
in Mol Cell (2020) Vol.78 pp811-813
Deep learning approach for quantification of organelles and misfolded polypeptides delivery within degradative compartments
Morone, D. Marazza, A. Bergmann, T. J. Molinari, M.
in Mol Biol Cell (2020) Vol.31 pp1512-1524
Mechanistic insights in recov-ER-phagy: micro-ER-phagy to recover from stress
Loi, M. Molinari, M.
in Autophagy (2020) Vol.16 pp385-386
ESCRT-III-driven piecemeal micro-ER-phagy remodels the ER during recovery from ER stress
Loi, M. Raimondi, A. Morone, D. Molinari, M.
in Nat Commun (2019) Vol.10 pp5058
Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways
Fregno, I. Molinari, M.
in Crit Rev Biochem Mol Biol (2019) Vol. 54, pp153-163
A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex
Forrester, A. De Leonibus, C. Grumati, P. Piemontese, M. Staiano, L. Fregno, I. Raimondi, A. Marazza, A. Bruno, G. Iavazzo, M. Intartaglia, D. Seczynska, M. van Anken, E. Conte, I. De Matteis, M. A. Dikic, I. Molinari, M. Settembre, C.
in EMBO J (2019) Vol.38 ppe99847
ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport
Fregno, I. Fasana, E. Bergmann, T. J. Loi, M. Solda, T. Galli, C. D’Antuono, R. Morone, D. Danieli, A. Paganetti, P. van Anken, E. Molinari, M.
in EMBO J (2018) Vol.37 ppe99259
Eat it right: ER-phagy and recovER-phagy
Loi, M. Fregno, I. Guerra, C.
in Biochem Soc Trans (2018) Vol. pp699–706
Endoplasmic reticulum turnover: ER-phagy and other flavors in selective and non-selective ER clearance
Fregno, I. Molinari, M.
in F1000Res (2018) Vol.7 pp454
Role of SEC62 in ER maintenance: A link with ER stress tolerance in SEC62-overexpressing tumors?
Bergmann, T. J. Fumagalli, F. Loi, M.
in Mol Cell Oncol (2017) Vol.4 ppe1264351
Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery
Fumagalli, F. Noack, J. Bergmann, T. J. Pisoni, G. B. Fasana, E. Fregno, I. Galli, C. Loi, M. Solda, T. D’Antuono, R. Raimondi, A. Jung, M. Melnyk, A. Schorr, S. Schreiber, A. Simonelli, L. Varani, L. Wilson-Zbinden, C. Zerbe, O. Hofmann, K. Peter, M. Quadroni, M. Zimmermann, R. Molinari, M.
in Nat Cell Biol (2016) Vol.18 pp1173-1184
Fumagalli, F. , Noack J. and Bergmann T. contributed equally to this work. * Recommended by the Faculty of 1000:
Researchers
Maurizio Molinari – Group Leader
Ilaria Fregno – Research Assistant
Tatiana Soldà – Scientist
Mikhail Rudinskiy – PhD student
Marco Fabbro – Master Student
Status: in progress
Overview
1) Aggregopathies (alpha1-antitrypsin deficiency)
The Z mutation of the SERPINA1 gene characterizes about 95% of the patients affected by alpha-1 antitrypsin deficiency (AATD), a lung and liver disease. The Z mutation results in a Glu342Lys substitution that encodes for alpha-1 antitrypsin-Z (ATZ). In about 10% of patients carrying the Z mutation, clearance of ATZ polymers from the ER is defective. This results in clinically significant hepatotoxicity, which is the major inherited cause of pediatric liver disease and transplantation. Notably, cell lines derived from AATD patients with liver disease (susceptible hosts) degrade ATZ and other polymerogenic serpins less efficiently than cells from AATD patients without liver disease (protected hosts). Our working hypothesis is that susceptible hosts have additional mutations leading to a loss-of-function in the pathway regulating clearance of ATZ polymers from cells. This would enhance accumulation of mutant ATZ within hepatocytes and make the patients more susceptible to liver injury. Our aim is to establish, in molecular detail the pathways that regulate clearance of ATZ polymers from mammalian cells using biochemical, cell biology, gene editing, optical and electron microscopy techniques. Identification of the gene products involved in clearance of ATZ polymers from our cells is expected to offer druggable targets to treat AATD and other human diseases resulting from misfolding and aggregation of mutant gene products.
Publications about aggregopathies
N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation
Fregno, I. Fasana, E. Solda, T. Galli, C. Molinari, M.
in EMBO J (2021) Vol. ppe107240
ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport
Fregno, I. Fasana, E. Bergmann, T. J. Loi, M. Solda, T. Galli, C. D’Antuono, R. Morone, D. Danieli, A. Paganetti, P. van Anken, E. Molinari, M.
in EMBO J (2018) Vol.37 ppe99259
Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways
Fregno, I. Molinari, M.
in Crit Rev Biochem Mol Biol (2019) Vol. 54, pp153-163.
2) Collagenopathies (spondyloepiphyseal dysplasia)
Collagens are the most abundant proteins in Metazoa, where they are major components of bones and cartilages. They are large polypeptide chains, which are prone to misfolding. Inherited or sporadic mutations in their sequence further reduce folding efficiency and are linked to rare diseases such as osteogenesis imperfecta, Ehlers-Danlos syndrome, Alport disease and many others. Solid scientific evidence is available, which shows that constitutive lysosomal clearance of defective collagen molecules maintains cellular and organ homeostasis. By studying the R989C mutant form of type II procollagen associated with spondyloepiphyseal dysplasia, we aim at identifying the cellular gene products involved in procollagen quality control. Defective quality control of newly synthesized proteins is linked to many rare and orphan disorders caused by protein misfolding, including collagenopathies. Identification of cellular factors promoting efficient folding and delivery of native proteins at their site of activity, and ensuring clearance from cells of polypeptides that cannot achieve their functional structure may offer druggable targets to treat such disorders.
Publications about collagenopathies
N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation
Fregno, I. Fasana, E. Solda, T. Galli, C. Molinari, M.
in EMBO J (2021) Vol. ppe107240
A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex
Forrester, A. De Leonibus, C. Grumati, P. Piemontese, M. Staiano, L. Fregno, I. Raimondi, A. Marazza, A. Bruno, G. Iavazzo, M. Intartaglia, D. Seczynska, M. van Anken, E. Conte, I. De Matteis, M. A. Dikic, I. Molinari, M. Settembre, C.
in EMBO J (2019) Vol.38 ppe99847
Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways
Fregno, I. Molinari, M.
in Crit Rev Biochem Mol Biol (2019) Vol. 54, pp153-163
3) Lysosomal storage disorders (Hunter’s syndrome, Gaucher disease, Morquio B, GBA1-linked Parkinson disease, GM1 gangliosidosis)
Lysosomal storage disorders are rare, inherited, progressive diseases characterized by impaired lysosomal activity with abnormal accumulation of macromolecules in the lumen of lysosomes. Our group has an ongoing collaboration with GAIN Therapeutics, a company founded in Ticino in 2017 and listed on the NASDAQ since March 2021 (https://www.gaintherapeutics.com/). GAIN Therapeutics exploits the computational platform SEE-Tx™ that uses the published 3D structure of enzymes and a proprietary computational technology to discover new allosteric binding sites and predict their druggability to correct enzyme misfolding, thus restoring function and eliminating the subsequent toxic substrate buildup that causes disease. Our group aims at understanding the mechanisms of action of compounds developed by GAIN to access allosteric sites in mutant enzymes thereby rescuing their lysosomal activity.
Publications about lisosomal storage disorders
Endoplasmic Reticulum and Lysosomal Quality Control of Four Nonsense Mutants of Iduronate 2-Sulfatase Linked to Hunter’s Syndrome
Marazza, A. Galli, C. Fasana, E. Burda, P. Fassi, E. M. A. Baumgartner, M. Cavalli, A. Molinari, M.
in DNA Cell Biol (2020) Vol.39 pp226-234
Researchers
Maurizio Molinari – Group Leader
Tatiana Soldà – Scientist
Status: In progress
Overview
The lumen of the ER contains 23 PDI members that insure formation of the correct set of intra- and inter-molecular disulfide bonds as a crucial, rate-limiting reaction of the protein folding process (Figure 1A). The reason for this high redundancy of PDIs remains unclear. Certainly, individual members of the PDI family show tissue-specific distribution or some kind of substrate preference (e.g. ERp57 forms functional complexes with the ER lectins calnexin and calreticulin and acts upon their ligands). The aim of this project is to uncover the role in protein biogenesis of the 5 type I membrane-bound members of the PDI family (TMX1, TMX2, TMX3, TMX4 and TMX5) (Figure 1B). Active PDIs contain the characteristic CXXC active-site motif that engages folding substrates in so-called mixed disulfides (i.e. covalent bonds between a PDI and a substrate cysteine). Mixed disulfides are extremely short living intermediates of the protein folding reaction, which can be stabilized upon replacement of the second (resolving) cysteine residue in the PDIs catalytic site. These so-called PDIs “trapping mutants” have been used to capture endogenous substrates of select ER-resident oxidoreductases such as ERp57, PDI, P5, ERp18, ERp72, ERp46 and ERdj5. The expression of a TMX1 trapping mutant in the living cells and the characterization by mass spectrometry of the polypeptides remaining covalently bound to it revealed a selective association with a series of cysteine-containing membrane-bound proteins. This is in contrast to studies performed with trapping mutants of other PDIs, which were all found to associate both with soluble and membrane-bound endogenous substrates. Studies are ongoing to confirm the substrate topology-dependent specificity of TMX1 and to characterize the role in protein biogenesis of the other TMX proteins (Figure 2).
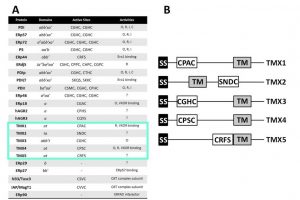
The PDI family. (A) The PDI family comprises 23 members. The 5 type I membrane-bound PDI family members (TMX proteins) are highlighted. (B) Domain structure of TMX1-TMX5.
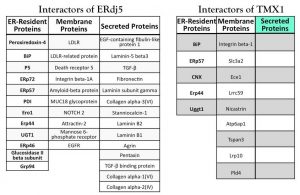
PDIs interactors. Endogenous proteins associated with the ERdj5 and TMX1 trapping mutant are shown.
Researchers
Maurizio Molinari – Group Leader
Elisa Fasana – Scientist
Ilaria Fregno – Scientist
Tatiana Soldà – Scientist
Status: In progress
Overview
Misfolded polypeptides produced in the ER are dislocated across the ER membrane to be degraded by cytosolic 26S-proteasomes in processes collectively defined as ERAD. Dislocation across the ER membrane is regulated by multimeric complexes built around one of the several membrane-embedded E3 ubiquitin ligases expressed in the mammalian ER. Physico-chemical features of the misfolded polypeptide (e.g. presence/absence of N-linked oligosaccharides, disulfide bonds, peptidyl-prolyl bonds in the cis conformation, membrane-anchor) may determine the quality control machineries that deliver the misfolded polypeptide at specific dislocation complexes. The definition of the rules that govern protein biogenesis and quality control requires a systematic analysis of appositely designed model folding-competent and folding-defective proteins. We have therefore prepared more than 50 model substrates with select physico-chemical features, whose fate will be monitored in mammalian cultured cells. The model polypeptides recapitulate structural defects found in mutant products of genes causing human disorders such as Alzheimer’s, Parkinson’s, Huntington’s diseases as well as many other rare genetic disorders characterized by gain-of-toxic-function or loss-of-function phenotypes. How the polypeptide’s features determine engagement of specific folding, quality control and degradation pathways will be determined in molecular details.
Publications
Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways
Fregno, I. Molinari, M.
in Crit Rev Biochem Mol Biol (2019) Vol. 54, pp153-163.
Researchers
Maurizio Molinari – Group Leader
Ilaria Fregno – Scientist
Elisa Fasana – Scientist
Carmela Galli Molinari – Scientist
Status: In progress
Autophagy is a conserved cellular process in eukaryotes required for degradation of cytoplasm contents into the lysosome/vacuole. Double-membrane vesicles called autophagosomes mediate the engulfment and transport of the cargo to be degraded during autophagy. While this pathway constitutively degrades cytoplasmic targets, it is also up-regulated by different cellular stresses. Starvation-induced autophagy randomly targets bulk cytoplasmic portions. Additionally, it selectively recognizes and degrades cytoplasmic protein aggregates, damaged organelles or invading microorganisms, playing thus a homeostatic and protective role in the cell. Interestingly, accumulation of misfolded proteins within the ER triggers autophagic degradation of portions of this organelle in yeast and mammals, suggesting that ER-phagy might be a conserved mechanism to prevent or overcome ER stress. While ERAD pathway is the classical and best characterized process for protein disposal in the ER, little is known about the mechanisms underlying ER degradation by autophagy. By using series of stable human cell lines created in our lab expressing regulated amounts of folding-competent and folding-defective protein chimeras, we are studying the contribution of autophagy in the degradation of these putative substrates and the molecular mechanisms regulating such a process. These studies will allow us to characterize the conditions for potential preferences in substrate elimination by ERAD and ER-phagy, and the mechanistic crosstalk between these two pathways and ER stress. The information generated by these studies will be validated in pathological model systems expressing disease-causing folding-defective proteins with the final goal of designing pharmacological treatments targeting protein disposal pathways to alleviate the toxicity caused by aberrant protein accumulation.
Publications
Proteasomal and lysosomal clearance of faulty secretory proteins: ER-associated degradation (ERAD) and ER-to-lysosome-associated degradation (ERLAD) pathways
Fregno, I. Molinari, M.
in Crit Rev Biochem Mol Biol (2019) Vol. 54, pp153-163
Deep learning approach for quantification of organelles and misfolded polypeptides delivery within degradative compartments
Morone, D. Marazza, A. Bergmann, T. J. Molinari, M.
in Mol Biol Cell (2020) Vol.31 pp1512-1524