
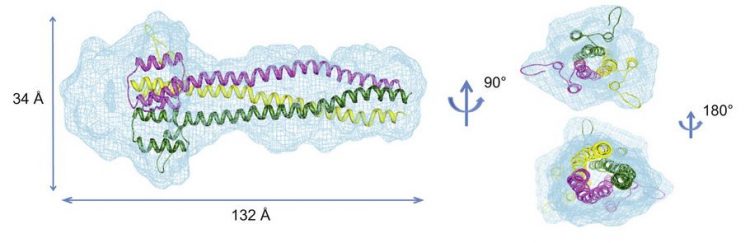
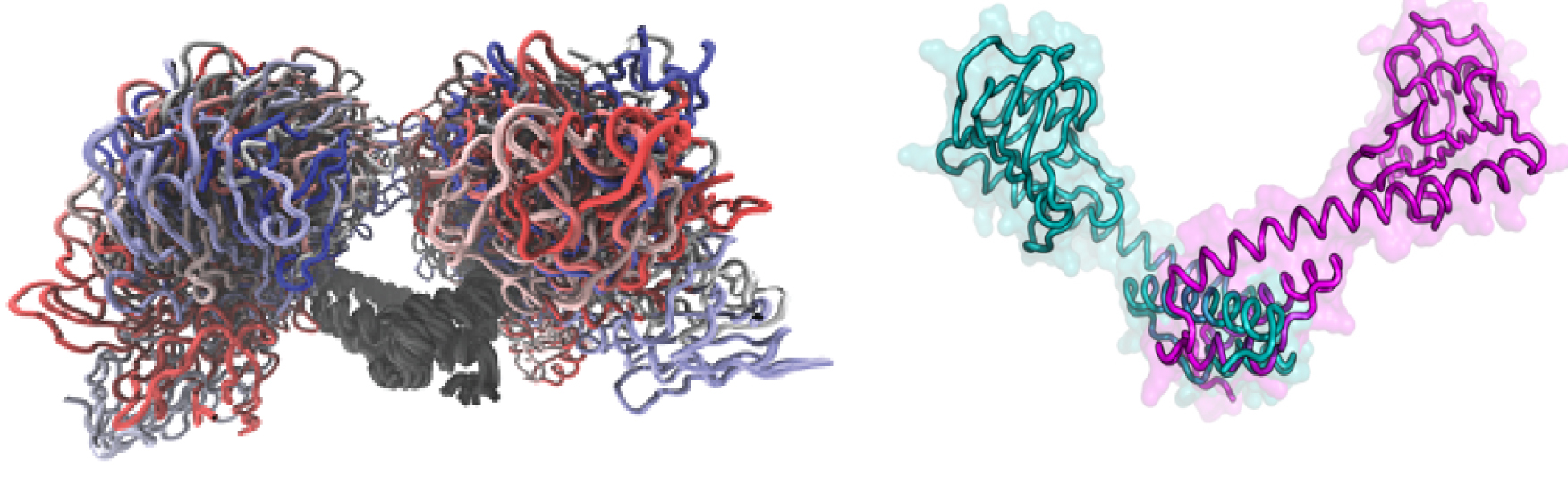
Nipah and Hendra viruses are recently emerged paramyxoviruses belonging to the Henipavirus genus. The Henipavirus phosphoprotein (P) consists of a large intrinsically disordered domain and a C-terminal domain (PCT) containing alternating disordered and ordered regions. Among these latter is the P multimerization domain (PMD). Using biochemical, analytical ultracentrifugation and small-angle X-ray scattering (SAXS) studies, we show that Hendra virus (HeV) PMD forms an elongated coiled-coil homotrimer in solution, in agreement with our previous findings on Nipah virus (NiV) PMD. However, the orientation of the N-terminal region differs from that observed in solution for NiV PMD, consistent with the ability of this region to adopt different conformations. SAXS studies provided evidence for a trimeric organization also in the case of PCT, thus extending and strengthening our findings on PMD. The present results are discussed in light of conflicting reports in the literature pointing to a tetrameric organization of paramyxoviral P proteins.
Signal Transducer and Activator of Transcription factors (STATs) are proteins able to translocate into the nucleus, bind DNA and activate gene transcription. STATs proteins play a crucial role in cell proliferation, apoptosis and differentiation. The prevalent view is that STATs proteins are able to form dimers and bind DNA only upon phosphorylation of specific tyrosine residues in the Trans-Activation-domain. However, this paradigm has been questioned recently by the observation of dimers of unphosphorylated STATs (USTATs) by X-ray, FRET and site-directed mutagenesis and a more complex view of the dimerization process and of the dimer role is emerging.
Due to its importance in cancer development and therapy, we focused our study on STAT3. Here we present an integrated modeling study in which we combine the available experimental data with different computational methodologies (homology modeling, protein-protein docking and molecular dynamics), to built reliable atomistic models of USTAT3 dimers. The models were validated performing computational alanine scanning for all the residues at the protein-protein interface. These results confirmed the experimental observations of the importance of some of these residues (in particular Leu78) USTAT3 dimerization process. Moreover, based on these models we were able to predict possible hot-spots (Gln32, Tyr79, Arg84, Arg93) for protein dimerization. In a future perspective our models could be valuable for understating the effects of important pathological mutations at molecular/atomistic level, and for in the design of new inhibitors of dimerization.
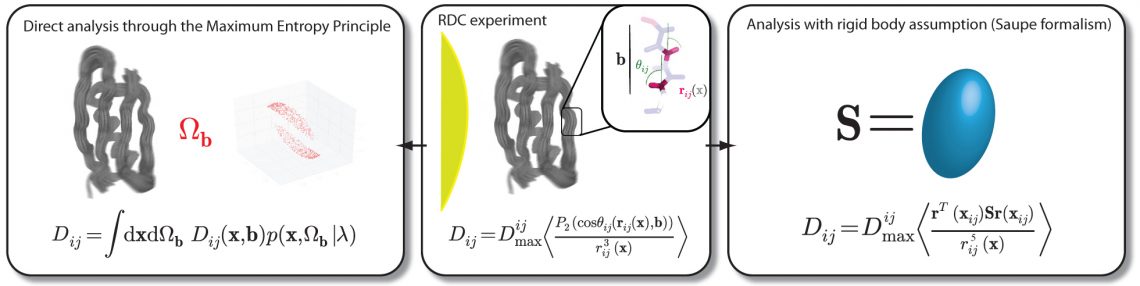
Olsson, S., et al. Molecular dynamics of biomolecules through direct analysis of dipolar couplings
Residual dipolar couplings (RDCs) are important probes in structural biology, but their analysis is often complicated by the determination of an alignment tensor or its associated assumptions. We here apply the Maximum Entropy principle to derive a tensor-free formalism that allows for direct, dynamic analysis of RDCs and holds the classic tensor formalism as a special case. Specifically, the framework enables us to robustly analyze data regardless of whether a clear separation of internal and overall dynamics is possible. Such a separation is often difficult in the core subjects of current structural biology, which include multi-domain and intrinsically disordered proteins as well as nucleic acids. We demonstrate the method is tractable, self-consistent and trivially generalizes to datasets comprised of observations from multiple different alignment conditions.